
《FDA安全与创新法案》修订低风险新医疗器械(de novo devic...
认证网 (2012/8/21 20:30:31) 浏览:2658 评论:0
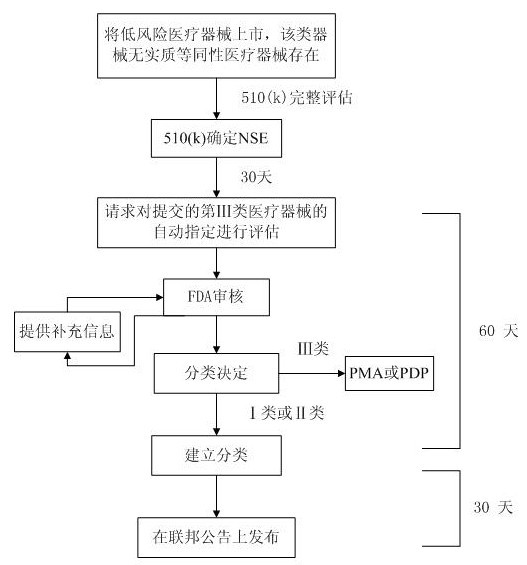
第Ⅲ类医疗器械的自动指定,也被称为“de novo”或“基于风险”分类,是指某类新的低风险类医疗器械因为没有与已经上市的医疗器械具有实质等同性而被划归为第Ⅲ类医疗器械,依据de novo机制将其重新划归为Ⅰ类或Ⅱ类器械的机制。在该机制下,当低风险新医疗器械申报者收到非实质等同性(NSE)决议的30天内,可以向FDA请求对申报的器械进行de novo分类。申报者须提供器械及详细信息的描述,并说明要求重新分类的理由。FDA会按照《联邦食品、药品和化妆品法》(FD&C Act)的513(a)(1)制定的要求和标准对器械进行重新分类。在申报者提交请求的60天内,FDA将会做出重新分类的决定,将申报的器械划归三种器械类型中的一种。第Ⅲ类医疗器械的自动指定评估的流程如图1所示:
图1:第Ⅲ类医疗器械自动指定评估的流程
* 在收到重新分类的要求时,FDA应在120天内依据FD&C Act的513(a)(1)中对三类器械的规定,对申请的器械进行分类;
* 当FDA发现一种合法上市的器械可以合理地判定实质性等同时,或者当FDA认为申报的器械并不是低-中等风险时,或者常规的控制并不足以控制风险并且不具备能够降低风险的特殊控制时,FDA可以拒绝接受重新分类的请求。
* 申报者在提交分类请求时,可以为其申报的器械推荐一种分类。如果推荐的分类为Ⅱ类控制,需在请求里包括一份可适用的特殊控制草案,特殊控制加上常规控制能够保证器械的安全和功效。并且,还应说明特殊控制是怎样保证器械的安全和功效。
图1:第Ⅲ类医疗器械自动指定评估的流程
* 在收到重新分类的要求时,FDA应在120天内依据FD&C Act的513(a)(1)中对三类器械的规定,对申请的器械进行分类;
* 当FDA发现一种合法上市的器械可以合理地判定实质性等同时,或者当FDA认为申报的器械并不是低-中等风险时,或者常规的控制并不足以控制风险并且不具备能够降低风险的特殊控制时,FDA可以拒绝接受重新分类的请求。
* 申报者在提交分类请求时,可以为其申报的器械推荐一种分类。如果推荐的分类为Ⅱ类控制,需在请求里包括一份可适用的特殊控制草案,特殊控制加上常规控制能够保证器械的安全和功效。并且,还应说明特殊控制是怎样保证器械的安全和功效。
检测认证热线:18768485300 & QQ在线服务:






