
医疗器械|美国市场|医疗器械分类
认证网 (2012/11/21 19:39:11) 浏览:979 评论:0
美国医疗器械的管理法规中对医疗器械产品建立了基于医学专业(用途)和基于产品风险的两种分类体系:
1. 基于医学专业(用途)的分类
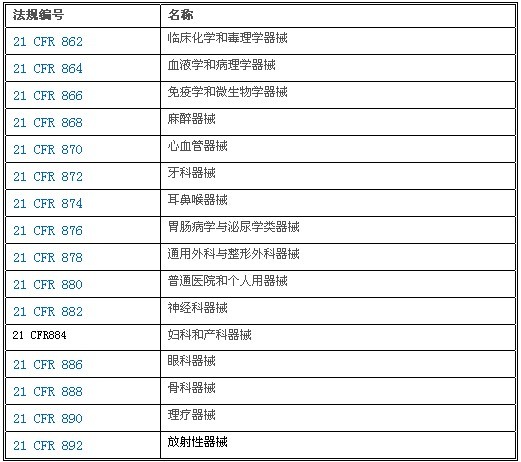
美国联邦行政法典21 CFR 862-892部分(见下表),将1700余种医疗器械产品结合医学专业划分为16大类,并给出了各种器械预期用途的相关描述。
2. 基于产品使用风险的管理分类
《联邦食品、药品和化妆品法》的513节(FD&C ACT section 513)中,根据医疗器械的使用风险和可能产生的危害程度,将其划分为三个管理类别,采取不同程度的控制措施。
Class I一般监管
这类器械只需经过一般监管就可以确保其功效与安全性,如拐杖、眼镜片、胶布等。一般监管的内容包括:禁止粗制滥造及不当标示的产品销售、不良产品限制或禁止销售和使用的规定、有关通知消费者和修理、更换、补偿金等售后服务的规定、GMP要求和制造商、进口商及分销商的企业注册与产品登记等。划入Class II及Class III的器械同样要遵守以上要求。
Class II一般监管和特别监管
II 类医疗器械是指单独依靠医疗器械的一般监管不足以确保医疗器械的安全性和有效性,而采取一些例如强制执行性能标准等特殊管理措施以提供相应的保证。此类产品包含医用手套、电动轮椅、助听器、血压计、诊疗导管等。II类器械除了遵守医疗器械的一般监管规定外,还须符合FDA所规定的特别要求或行业公认的标准等。FDA的特别要求中,包括了对特定产品的强制性性能标准(mandatory performance standards)、特殊标识要求以及上市后监督等。
Class III 一般监管和上市前批准
被列入Class III的产品多为维持、支持生命或植入体内的器械,它们对病人具有潜在危险,可能引发伤害或疾病。这类产品包括心律调节器、子宫内械及婴儿保温箱等。除一般监管的规定外,美国通过上市前批准(PMA)制度设置了一系列科学的审核程序,作为确保III类器械安全和有效的一种必要的市场准入措施。但是,并非所有的III类器械都需要通过PMA的方式获得市场准入许可。目前,仅有约20%的III类器械必须取得PMA方能进入美国市场,其他III类器械可依据CFR法规通过[510(k)]申请等方式获得上市许可。
基于风险的分类制度是FDA从实施有效监管的角度出发而建立的。产品被列入不同的管理类别,意味着其制造商向FDA申请获准上市的程序和提交资料将有所区别。所有I类和 II类器械(享受的豁免的除外)均须按照FD&C ACT 510(k)条款的要求执行上市前通告(Premarket Notification),向FDA申请产品上市许可;所有III类器械(除在1976年《医疗器械修正案》通过(1976年5月28日)前上市的器械(preamendments device)及其等价器械外),必须按照PMA的要求申请上市许可。
联邦食品、药品和化妆品法513节中规定,医疗器械分类工作由分类专家小组负责完成。目前,FDA根据依用途划分的16大类器械而设置了16个相应的分类专家小组,每个小组由一位该领域的医学专家和六位学者所组成,小组在讨论某一医疗器械产品分类的过程中允许不具投票权的消费者代表、行业及企业代表参与。对于器械的重新分类问题,除了FDA主动重新分类之外,制造商也可向FDA提出重新分类的申请。
1. 基于医学专业(用途)的分类
美国联邦行政法典21 CFR 862-892部分(见下表),将1700余种医疗器械产品结合医学专业划分为16大类,并给出了各种器械预期用途的相关描述。
2. 基于产品使用风险的管理分类
《联邦食品、药品和化妆品法》的513节(FD&C ACT section 513)中,根据医疗器械的使用风险和可能产生的危害程度,将其划分为三个管理类别,采取不同程度的控制措施。
Class I一般监管
这类器械只需经过一般监管就可以确保其功效与安全性,如拐杖、眼镜片、胶布等。一般监管的内容包括:禁止粗制滥造及不当标示的产品销售、不良产品限制或禁止销售和使用的规定、有关通知消费者和修理、更换、补偿金等售后服务的规定、GMP要求和制造商、进口商及分销商的企业注册与产品登记等。划入Class II及Class III的器械同样要遵守以上要求。
Class II一般监管和特别监管
II 类医疗器械是指单独依靠医疗器械的一般监管不足以确保医疗器械的安全性和有效性,而采取一些例如强制执行性能标准等特殊管理措施以提供相应的保证。此类产品包含医用手套、电动轮椅、助听器、血压计、诊疗导管等。II类器械除了遵守医疗器械的一般监管规定外,还须符合FDA所规定的特别要求或行业公认的标准等。FDA的特别要求中,包括了对特定产品的强制性性能标准(mandatory performance standards)、特殊标识要求以及上市后监督等。
Class III 一般监管和上市前批准
被列入Class III的产品多为维持、支持生命或植入体内的器械,它们对病人具有潜在危险,可能引发伤害或疾病。这类产品包括心律调节器、子宫内械及婴儿保温箱等。除一般监管的规定外,美国通过上市前批准(PMA)制度设置了一系列科学的审核程序,作为确保III类器械安全和有效的一种必要的市场准入措施。但是,并非所有的III类器械都需要通过PMA的方式获得市场准入许可。目前,仅有约20%的III类器械必须取得PMA方能进入美国市场,其他III类器械可依据CFR法规通过[510(k)]申请等方式获得上市许可。
基于风险的分类制度是FDA从实施有效监管的角度出发而建立的。产品被列入不同的管理类别,意味着其制造商向FDA申请获准上市的程序和提交资料将有所区别。所有I类和 II类器械(享受的豁免的除外)均须按照FD&C ACT 510(k)条款的要求执行上市前通告(Premarket Notification),向FDA申请产品上市许可;所有III类器械(除在1976年《医疗器械修正案》通过(1976年5月28日)前上市的器械(preamendments device)及其等价器械外),必须按照PMA的要求申请上市许可。
联邦食品、药品和化妆品法513节中规定,医疗器械分类工作由分类专家小组负责完成。目前,FDA根据依用途划分的16大类器械而设置了16个相应的分类专家小组,每个小组由一位该领域的医学专家和六位学者所组成,小组在讨论某一医疗器械产品分类的过程中允许不具投票权的消费者代表、行业及企业代表参与。对于器械的重新分类问题,除了FDA主动重新分类之外,制造商也可向FDA提出重新分类的申请。
检测认证热线:18768485300 & QQ在线服务:
相关内容:






