
医疗器械|欧盟市场|监管法规体系
认证网 (2012/11/21 19:15:32) 浏览:1332 评论:0
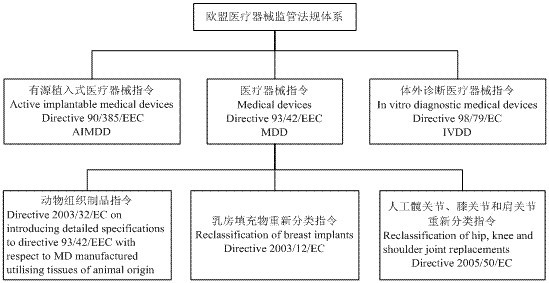
欧盟现行的医疗器械监管法规体系如下图所示:
有源植入式医疗器械指令(Active Implantable Medical Devices Directive 90/358/EC,AIMDD),覆盖所有有源植入式医疗器械产品,例如,心脏起搏器、体内给药器械产品、除纤颤器等。该指令于1993年被93/42/EEC指令和93/68/EEC指令两度修订;又于2007年被2007/47/EC指令进行了第三次修订;
体外诊断医疗器械指令(In Vitro Diagnostic Directive 98/79/EEC,IVDD),覆盖所有体外诊断试剂和仪器。2002年,欧盟委员会发布2002/364/EC决定,作为实施措施对该指令进行了修订;
医疗器械指令(Medical Devices Directive 93/42/EEC,MDD),覆盖除有源植入式医疗器械和体外诊断医疗器械以外的所有用于人体的医疗器械及其附件,包括无源植入物、外科器械、医疗电子设备等。该指令自发布之日起,分别被98/79/EC指令、2000/70/EC指令、2001/104/EC指令、No.1882/2003法规和2007/47/EC指令先后进行了五次修订;
动物组织制品指令(2003/32/EC),涉及所有动物源医疗器械产品。要求该类产品生产企业必须按照指令中的原则实施合适的且被证明了的风险分析与风险管理策略,采取措施将传染风险降至最低,将残留风险控制在人体可接受范围。使用动物组织生产的医疗设备,只有同时符合动物组织制品指令(2003/32/EC)和医疗器械指令(93/42/EEC),该产品才能在欧洲市场上销售;
乳房植充物重新分类指令(2003/12/EC),根据医疗器械指令(93/42/EEC)附录IX的分类规则,乳房填充物所属的管理类别由原来的IIb类调整为III类;
人工髋关节、膝关节和肩关节重新分类指令(2005/50/EC),根据医疗器械指令(93/42/EEC)附录IX的分类规则,人工髋关节、膝关节和肩关节所属的管理类别由原来的IIb类调整为III类。
从欧盟医疗器械监管法规体系的构成来看,产品范围成为技术法规制定的出发点。有源植入式器械和体外诊断器械因其特殊性,被欧盟以专门立法的形式实施监管。虽然管理的产品范围有所区别,但AIMDD、IVDD和MDD三个指令在管理内容方面却体现出统一性,管理措施与要求均涵盖了产品上市前和上市后两个阶段。
为了不断提高医疗器械的安全性,欧盟对AIMDD、IVDD和MDD分别进行了若干次修订。2007年9月,2007/47/EC指令的发布,被业界认为是十四年来欧盟对医疗器械指令(93/42/EEC)最为重大的一次修订,从简单的文字修正到新要求的引入,共涉及150余处变化。另外,该指令同时还修订了有源植入式医疗器械指令(90/358/EC)的部分条款。根据这一指令的规定,欧盟各成员国应在2008年12月21日前完成将指令转化为本国法规的工作,新指令定于2010年3月21日正式开始实施。鉴于此,国内广大出口欧盟的医疗器械生产企业应及时和掌握新的法规变化,提前采取措施积极应对。
根据以上医疗器械法规,所有计划进入欧盟市场的医疗器械产品在上市前必须实施指令规定的适用的合格评定程序。欧盟为保证这些合格评定程序的有效性,从各成员国的第三方合格评定机构中统一认定了一批“公告机构”(Notified Body,NB),负责参与除I类器械外器械的合格评定过程,授权其对产品和制造商的质量体系/产品设计检验实施上市前审查,为审查合格的产品颁发质量体系批准证书/EC设计检验证书。产品在获得相关证书后即可加贴CE标志,实现在欧盟市场的顺利流通。欧盟同时要求所在国主管部门对公告机构进行监督,定期检查其审查工作的执行情况和财务状况,以确保公告机构能够秉公执法。此外,欧盟还定期召开各国管理部门会议,讨论、交流对公告机构的监督情况。公告机构也致力于统一审查方法和标准,以便减少不同机构对同一类别产品审查时的差异。
有源植入式医疗器械指令(Active Implantable Medical Devices Directive 90/358/EC,AIMDD),覆盖所有有源植入式医疗器械产品,例如,心脏起搏器、体内给药器械产品、除纤颤器等。该指令于1993年被93/42/EEC指令和93/68/EEC指令两度修订;又于2007年被2007/47/EC指令进行了第三次修订;
体外诊断医疗器械指令(In Vitro Diagnostic Directive 98/79/EEC,IVDD),覆盖所有体外诊断试剂和仪器。2002年,欧盟委员会发布2002/364/EC决定,作为实施措施对该指令进行了修订;
医疗器械指令(Medical Devices Directive 93/42/EEC,MDD),覆盖除有源植入式医疗器械和体外诊断医疗器械以外的所有用于人体的医疗器械及其附件,包括无源植入物、外科器械、医疗电子设备等。该指令自发布之日起,分别被98/79/EC指令、2000/70/EC指令、2001/104/EC指令、No.1882/2003法规和2007/47/EC指令先后进行了五次修订;
动物组织制品指令(2003/32/EC),涉及所有动物源医疗器械产品。要求该类产品生产企业必须按照指令中的原则实施合适的且被证明了的风险分析与风险管理策略,采取措施将传染风险降至最低,将残留风险控制在人体可接受范围。使用动物组织生产的医疗设备,只有同时符合动物组织制品指令(2003/32/EC)和医疗器械指令(93/42/EEC),该产品才能在欧洲市场上销售;
乳房植充物重新分类指令(2003/12/EC),根据医疗器械指令(93/42/EEC)附录IX的分类规则,乳房填充物所属的管理类别由原来的IIb类调整为III类;
人工髋关节、膝关节和肩关节重新分类指令(2005/50/EC),根据医疗器械指令(93/42/EEC)附录IX的分类规则,人工髋关节、膝关节和肩关节所属的管理类别由原来的IIb类调整为III类。
从欧盟医疗器械监管法规体系的构成来看,产品范围成为技术法规制定的出发点。有源植入式器械和体外诊断器械因其特殊性,被欧盟以专门立法的形式实施监管。虽然管理的产品范围有所区别,但AIMDD、IVDD和MDD三个指令在管理内容方面却体现出统一性,管理措施与要求均涵盖了产品上市前和上市后两个阶段。
为了不断提高医疗器械的安全性,欧盟对AIMDD、IVDD和MDD分别进行了若干次修订。2007年9月,2007/47/EC指令的发布,被业界认为是十四年来欧盟对医疗器械指令(93/42/EEC)最为重大的一次修订,从简单的文字修正到新要求的引入,共涉及150余处变化。另外,该指令同时还修订了有源植入式医疗器械指令(90/358/EC)的部分条款。根据这一指令的规定,欧盟各成员国应在2008年12月21日前完成将指令转化为本国法规的工作,新指令定于2010年3月21日正式开始实施。鉴于此,国内广大出口欧盟的医疗器械生产企业应及时和掌握新的法规变化,提前采取措施积极应对。
根据以上医疗器械法规,所有计划进入欧盟市场的医疗器械产品在上市前必须实施指令规定的适用的合格评定程序。欧盟为保证这些合格评定程序的有效性,从各成员国的第三方合格评定机构中统一认定了一批“公告机构”(Notified Body,NB),负责参与除I类器械外器械的合格评定过程,授权其对产品和制造商的质量体系/产品设计检验实施上市前审查,为审查合格的产品颁发质量体系批准证书/EC设计检验证书。产品在获得相关证书后即可加贴CE标志,实现在欧盟市场的顺利流通。欧盟同时要求所在国主管部门对公告机构进行监督,定期检查其审查工作的执行情况和财务状况,以确保公告机构能够秉公执法。此外,欧盟还定期召开各国管理部门会议,讨论、交流对公告机构的监督情况。公告机构也致力于统一审查方法和标准,以便减少不同机构对同一类别产品审查时的差异。
检测认证热线:18768485300 & QQ在线服务:
相关内容:






